Center for Motor Neuron Biology and Disease

Motor Neuron Center
Columbia’s Motor Neuron Center will transform our understanding of human health. For the first time, brilliant scientific minds are working together in a common approach to currently incurable motor neuron diseases: spinal muscular atrophy (SMA) in children and amyotrophic lateral sclerosis (ALS; Lou Gehrig’s disease) in adults. New discoveries in the field of motor neuron biology will fuel the search for effective therapy for patients.

News
-
Haploinsufficiency of the SLC2A1 gene and paucity of its translated product, the glucose transporter-1 (Glut1) protein, disrupt brain function and cause the neurodevelopmental disorder...
-
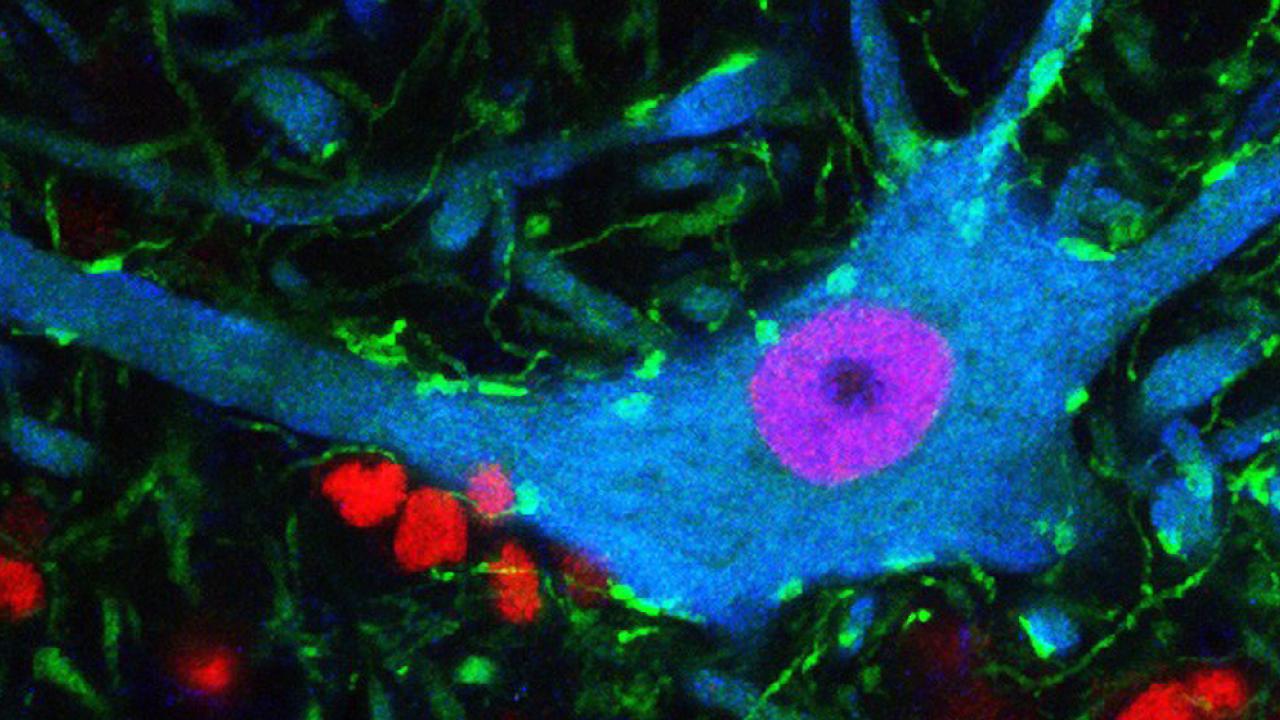
Behavioral deficits in neurodegenerative diseases are often attributed to the selective dysfunction of vulnerable neurons via cell-autonomous mechanisms.
Topic
Research -
The RNA-binding protein FUS participates in several RNA biosynthetic processes and has been linked to the pathogenesis of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia.
Topic
Research
Support the Center

Your support of Columbia’s Motor Neuron Center will benefit patients and their families through earlier diagnosis, improved patient care, and the discovery of novel treatments.